 Sebé-Pedrós Lab
Sebé-Pedrós Lab
Systems and Synthetic Biology
- Group page
- Research lines
- Group members
- Publications


![]()
2009-2013 - PhD in Genetics. University of Barcelona (UB). Spain.
2013-2015 - Research associate. Institute for Evolutionary Biology (CSIC-UPF). Spain
2015-2018 - EMBO/WIS Postdoctoral Fellow. Weizmann Institute of Science (WIS). Israel
2019 - Group Leader at the Centre for Genomic Regulation (CRG). Spain
News
First Steps to Launch Ambitious Quest to Chart Cellular Biodiversity (08/01/2024)
To understand biodiversity at the cellular level, researchers have to create detailed maps of all the different types of cells in many different organisms, also known as a cell atlas.
Tiny sea creatures reveal the ancient origins of neurons (19/09/23)
A study in the journal Cell sheds new light on the evolution of neurons, focusing on the placozoans, a millimetre-sized marine animal.
CRG researchers selected for EMBO Young Investigator programme (22/11/2022)
CRG researchers Lars Velten and Arnau Sebe Pedros are two of 24 life scientists to be selected for this year’s EMBO Young Investigator Programme.
Chromatin originated in ancient microbes one to two billion years ago (09/06/2022)
Researchers at the CRG reveal that nature’s storage solution first evolved in ancient microbes living on Earth between one and two billion years ago.
Researchers create critical new resource to boost coral reef conservation efforts (03/05/2021)
The findings provide new insights into the molecular biology and evolution of the reef-building stony coral
CRG scientists receive €5m to research cancer, ageing and evolution (03/09/2019)
Arnau Sebé-Pedrós, Group Leader in the Systems Biology research programme, has received funding for EvoCellMap, a project tracing the origin and evolution of animal cell types.
Summary
Sebé-Pedrós lab website: https://www.sebepedroslab.org/
A fundamental question in biology is how the diverse cell types observed in a multicellular organism are encoded by a single genome sequence, and which genome regulatory mechanisms orchestrate the deployment and maintenance of cell type-specific transcriptional programs. However, the diversity and evolutionary dynamics of cell type programs remains almost unexplored beyond selected tissues in a few species. Similarly, little is known about the emergence of complex genome regulatory mechanisms that support cell type-specific programs and cellular memory, for example genome spatial compartmentalization and repressive chromatin modifications.
In recent years, the development of advanced functional genomics technologies has revolutionized the study of cell type and genome regulation, even at single-cell resolution. This opens the way to the comparative analysis of genome regulation in species that represent diverse levels of biological complexity: ranging from unicellular temporal differentiation and simple multicellular behaviours (e.g. in some protistan eukaryotes), through loosely integrated and limitedly diversified ensembles of cell types (e.g. in early-branching animals), to organisms with elaborate tissue and bodyplan organization (e.g in bilaterian animals).
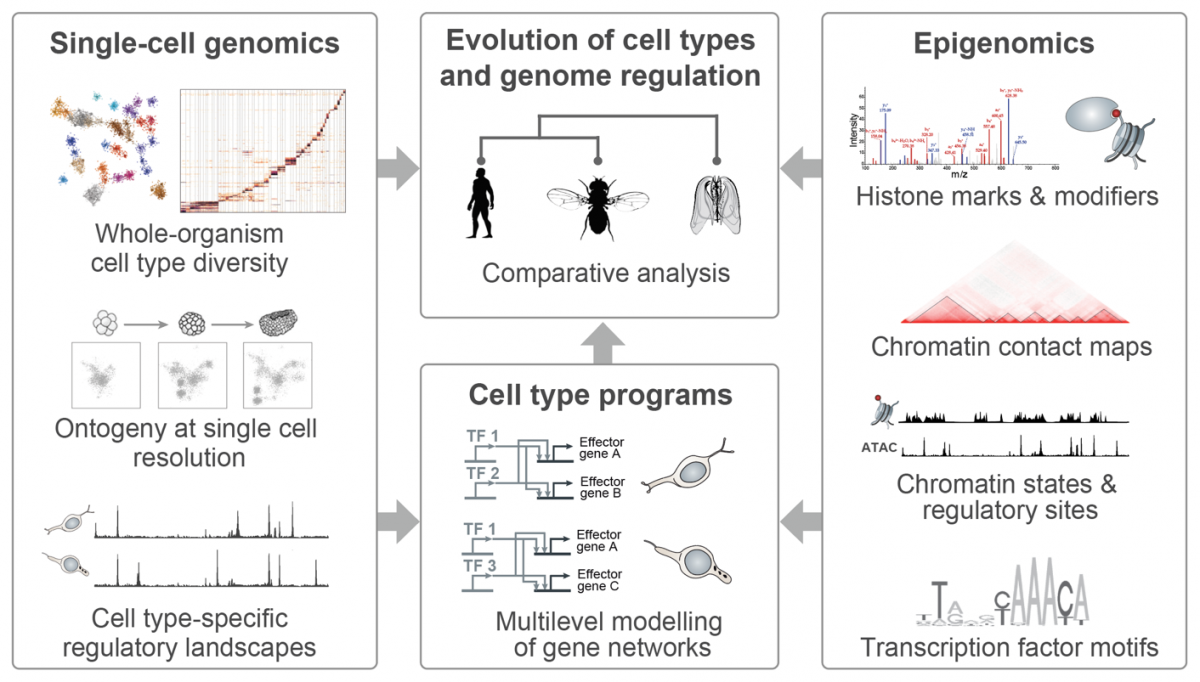
|
(Illustrations adapted from Sebé-Pedrós et al., Cell 2016, Sebé-Pedrós et al. Cell 2018, Arendt et al. NatRevGen 2016, Bonev & Cavalli, NatRevGen 2016, and Booth & King Nature 2016) |
In our group, we combine high-throughput epigenomics and single-cell genomics technologies with advanced computational methods in order to dissect cell type programs and genome regulatory architectures in phylogenetically diverse systems. The comparative analysis of these data allows us (i) to trace the evolution of cell types and genome regulatory mechanisms; (ii) to identify shared principles in genome function; and (iii) to reconstruct regulatory innovations linked to major transitions such as the origin of eukaryotic cells or the emergence of multicellular organisms.
Funding Acknowledgements

The project “Desarrollo de modelos cuantitativos para entender la evolución de las redes génicas de identidad celular en animales” (PID2021-124757NB-I00) is funded by Agencia Estatal de Investigación (AEI), the Ministerio de Ciencia e Innovación and Fondo Europeo de Desarrollo Regional (FEDER). Project PID2021-124757NB-I00 funded by MCIN/ AEI / 10.13039/501100011033 / FEDER, UE
1. Single-cell genomics of cell type diversity
Cell types are the basic building blocks of multicellular animals. Traditional studies based on microscopy and spatial gene expression profiling (by in situ hybridization) have provided important insights into the evolution of specific cell types in different lineages. However, these approaches require a priori selection of gene markers; they are difficult to scale towards multiple expressed genes simultaneously; and they are not readily applicable to all species or life stages, in particular adult specimens. As a consequence, we lack a systematic and unbiased understanding of the diversity and evolution of animal cell types.
In this research line, we apply and develop single-cell transcriptomics tools to unbiasedly characterize and compare cell types in whole-adult organisms and embryonic stages of diverse animal lineages.
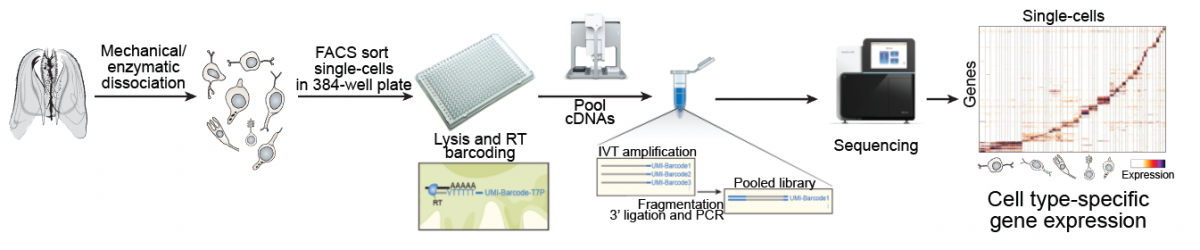
|
Whole-organism cell type diversity by single-cell RNA-seq. |
2. Comparative modelling of cell type gene regulatory networks
The implementation of cell type-specific transcriptional programs is controlled by gene regulatory networks (GRNs), in which key elements are transcription factors (TFs) and the cis-regulatory sites where these TFs bind. Therefore, changes in the structure and composition of these GRNs are likely to underlie cell type evolution. Moreover, similarities in cell type-specific TF regulators and GRNs are indicators of cell type homology. However, the systematic analysis of GRNs within and between species has been precluded by several technical constraints, for example the absence of specific ChIP antibodies to profile the binding sites of orthologous TFs in different species.
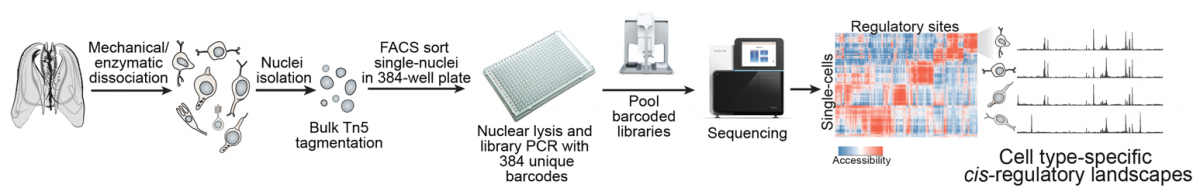
|
Single-cell chromatin accessibility to reconstruct cell type cis-regulatory landscapes. |
| To by-pass these limitations, in our group we integrate cell type-specific expression and chromatin data with computational inference methods (e.g. TF sequence motif analysis) in order to model the GRNs underlying cell type hierarchies in different metazoan species. Our ultimate goal is to compare these genetic programs in order to (i) reconstruct the structure and evolution of specific GRNs (e.g. neuronal networks), (ii) define general principles of GRN evolution in metazoans, and (iii) use similarities in the usage of orthologous TFs and their networks to assess cell type homologies across species. |
|
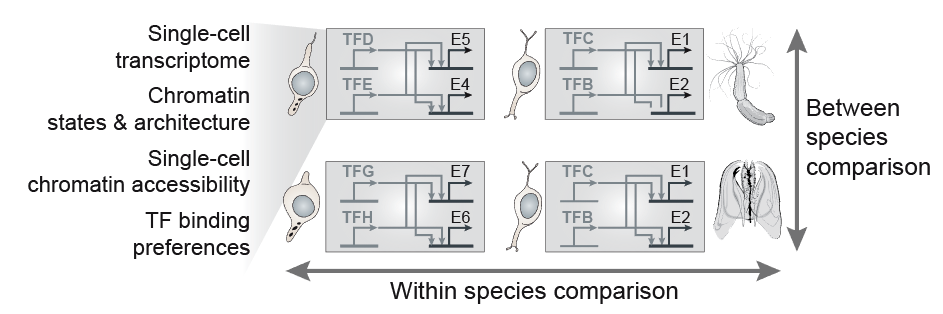
3. Evolution of chromatin regulation and genome architecture
|
The access to eukaryotic genetic information is controlled by a complex nucleoproteic interface called chromatin. Besides transcription factors, the other major trans components of the chromatin include histone proteins and associated readers, writers and erasers of histone post-translational modifications (hPTMs); as well as proteins that mediate chromatin folding (e.g. CTCF and cohesins). Together, these chromatin processes have a crucial role in the establishment and maintenance of cell identity. However, to date most research in chromatin and genome regulatory biology has focused in a very small set of organisms, including model animals (mouse, Drosophila and C.elegans), plants (Arabidopsis, Zea), and model unicellular fungi (Saccharomyces, Schizosaccharomyes), as well as human parasites: Plasmodium (Alveolata), Toxoplasma (Alveolata) and Trypanosoma (Excavata).
|
|
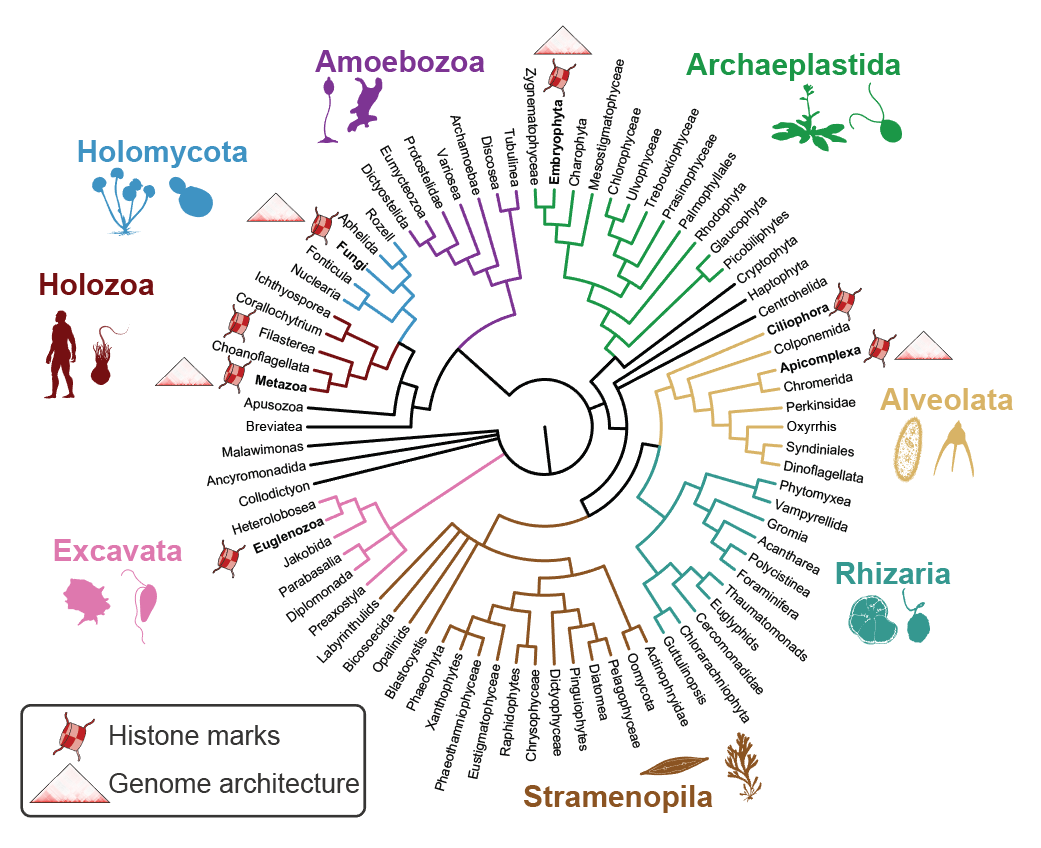
In this research line, we aim at expanding our understanding of genome regulatory mechanisms from a phylogenetic perspective. To this end, we apply high-throughput chromosomal conformation capture (HiC) and chromatin immunoprecipitation (ChIP-seq) analysis to define and compare the regulatory genome architectures of unicellular and multicellular eukaryotes at key phylogenetic positions. Similarly, using comparative genomics and proteomics analyses, we study the origin and evolution of eukaryotic transcription factors, histone PTMs and their interactome of readers and modifiers.

Group Leader
Postdoctoral Researchers
PhD Students
Technicians
Postdoctoral Researchers
PhD Students
Technicians
![]()

![]()